
About Ferret
Ferret is a user-friendly tool to quickly extract human genetic variation data from the latest release of the 1000 Genomes (1KG) Project. Ferret was developed as a straightforward Java application to be accessible even for non-specialists who are not adept at bioinformatics. By converting the 1KG vcf files to a format that can be read by popular pre-existing tools (e.g. Plink and HaploView), Ferret offers easy manipulation and visualization of the 1KG SNP and indel data, easy access to allelic frequency, linkage disequilibrium and haplotype information, and eventually tagSNP design.
What does Ferret do?
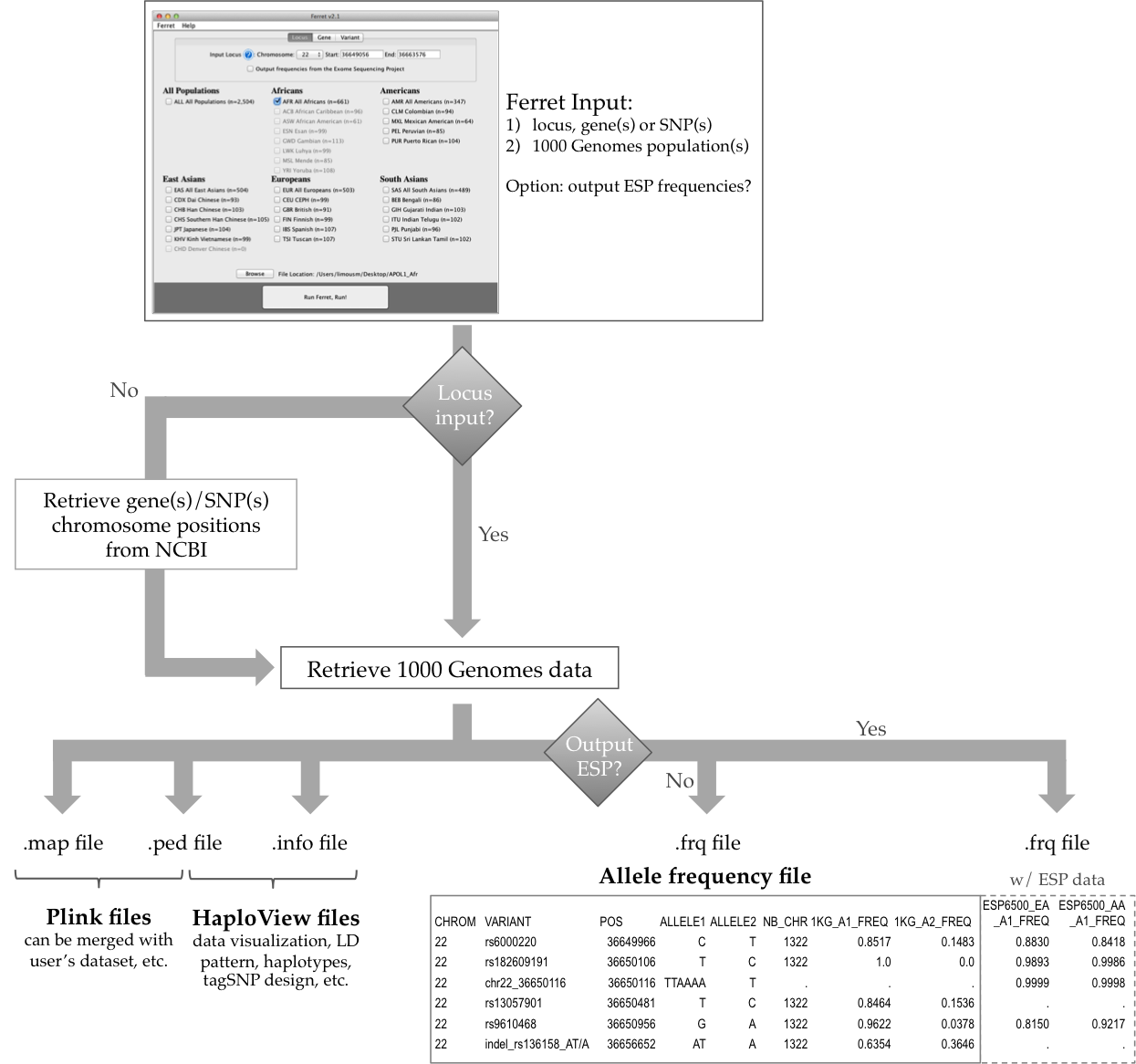
*1KG, 1000 Genomes; ESP, Exome Sequencing Project; LD, linkage disequilibrium; CHROM, chromosome; POS, position; NB_CHR, number of chromosomes; A1, allele 1; A2, allele 2; FREQ, frequency.
Ferret unique features:
- Can extract data from a locus, from a gene or gene list, or from a variant ID or a variant list
- Converts 1KG SNP and indel data
- Computes allelic frequency for 1KG SNP, indel and CNV variants
- Retrieves allelic frequency for the Exome Sequencing Project variants
- Creates output files suitable for popular pre-existing tools (e.g. Plink and HaploView)
- User-friendly interface
Ferret options
The user can access the Settings panel in the Ferret tab and can set the following options:
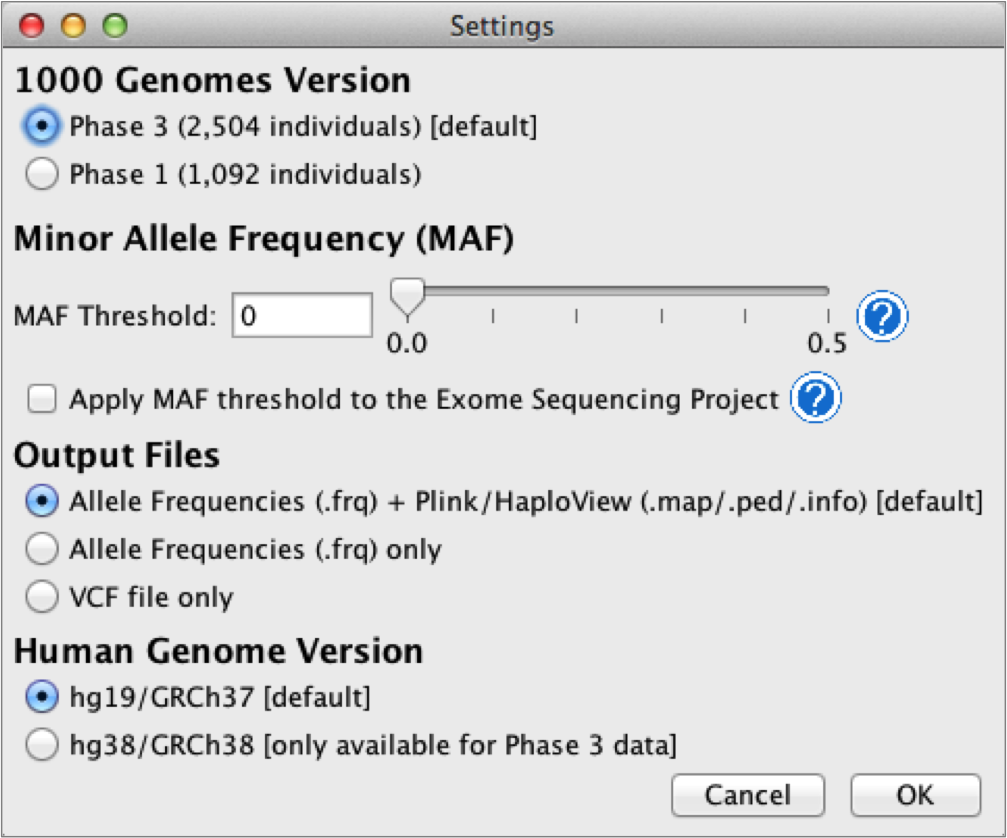
- 1000 Genomes version: Phase 3 (default) or Phase 1
- Human genome version: hg19/GRCh37 (default) or hg38/GRCh38
- Minor allele frequency (MAF) threshold for variants to output
- Output file format: Plink/HaploView files + allele frequency file, or allele frequency file only, or .vcf file
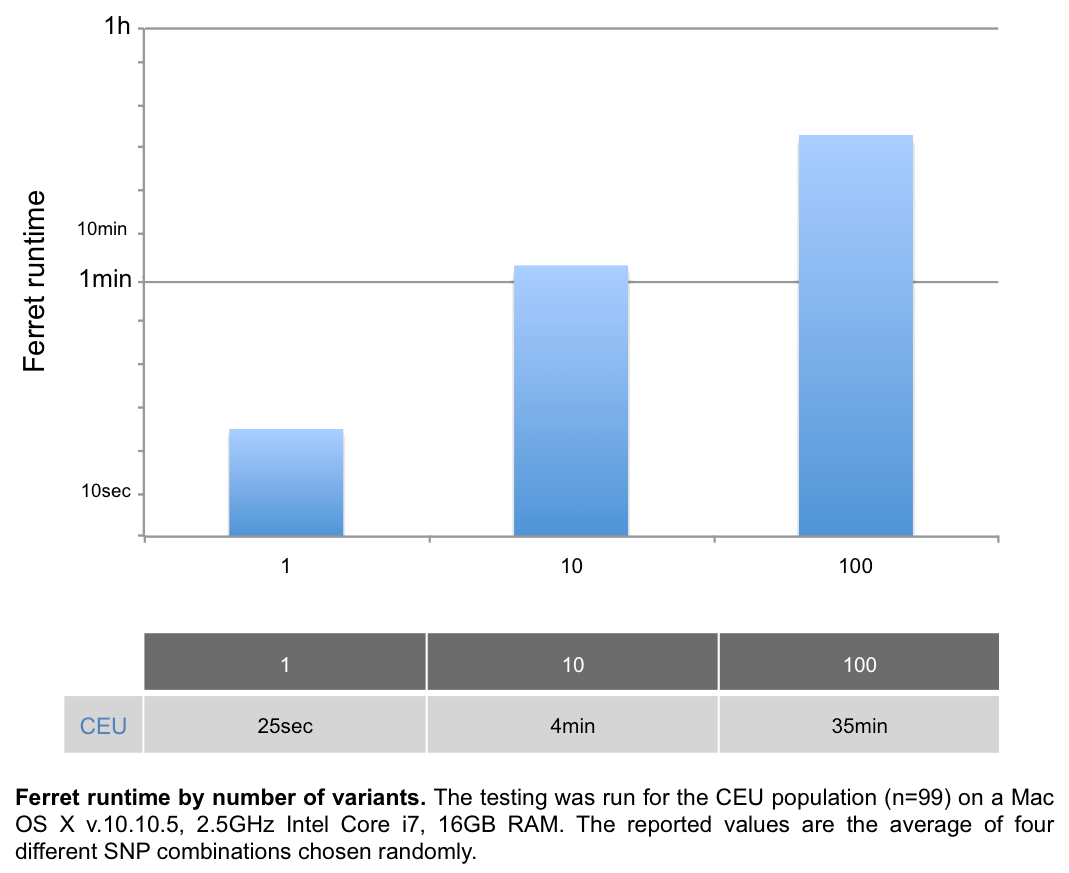
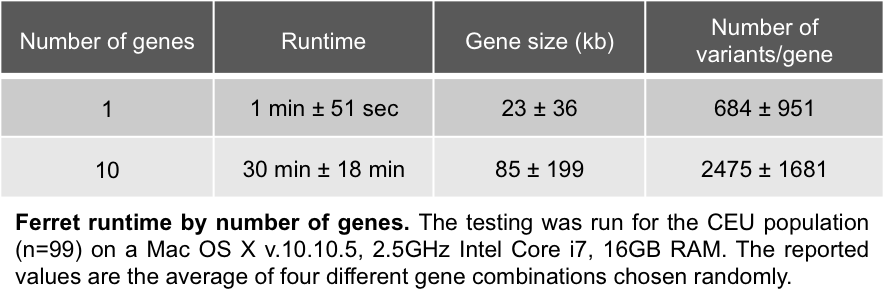
Ferret runtime
Several factors can impact runtime, such as the computer specs, the network quality, or the 1KG server access (e.g. running a large query at 2pm EST takes longer than at 9pm EST), so the numbers below are given for guidance only. The population size does not significantly impact Ferret runtimes.
- For locus inputs, Ferret runs for about 3min for a 100kb locus and 35min for a 1Mb locus:
- For SNP inputs, Ferret runtime is approximately 4min for 10 SNPs and 35min for 100 SNPs:
- Runtimes are much more variable for gene inputs (between 16 and 55 min for 10 genes), as gene size and variant density can be highly variable:
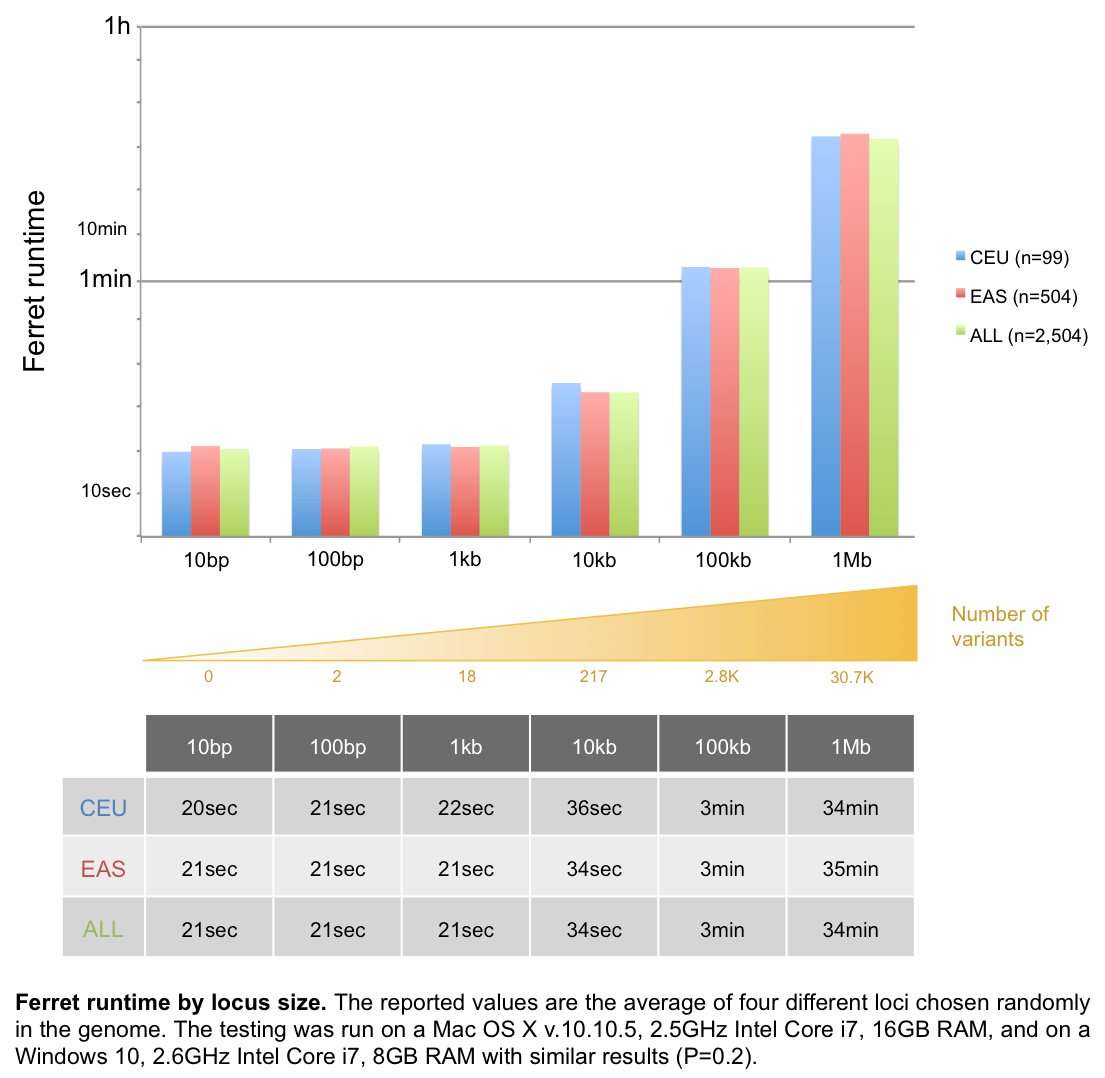
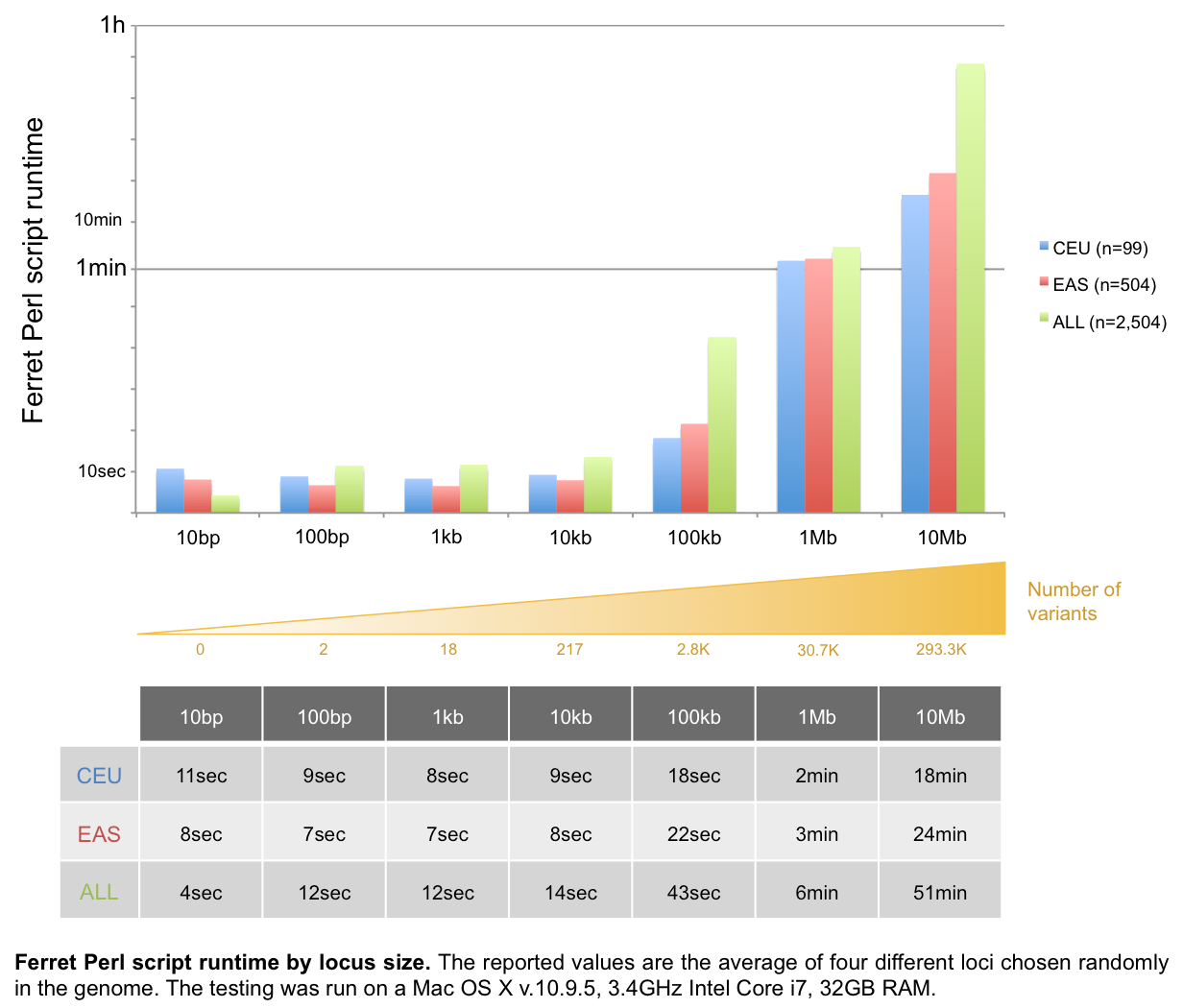
For locus with a size ≥1Mb, we recommend using our Perl script, which is faster than our Java application (P=2.7x10-4).
{kind=link}
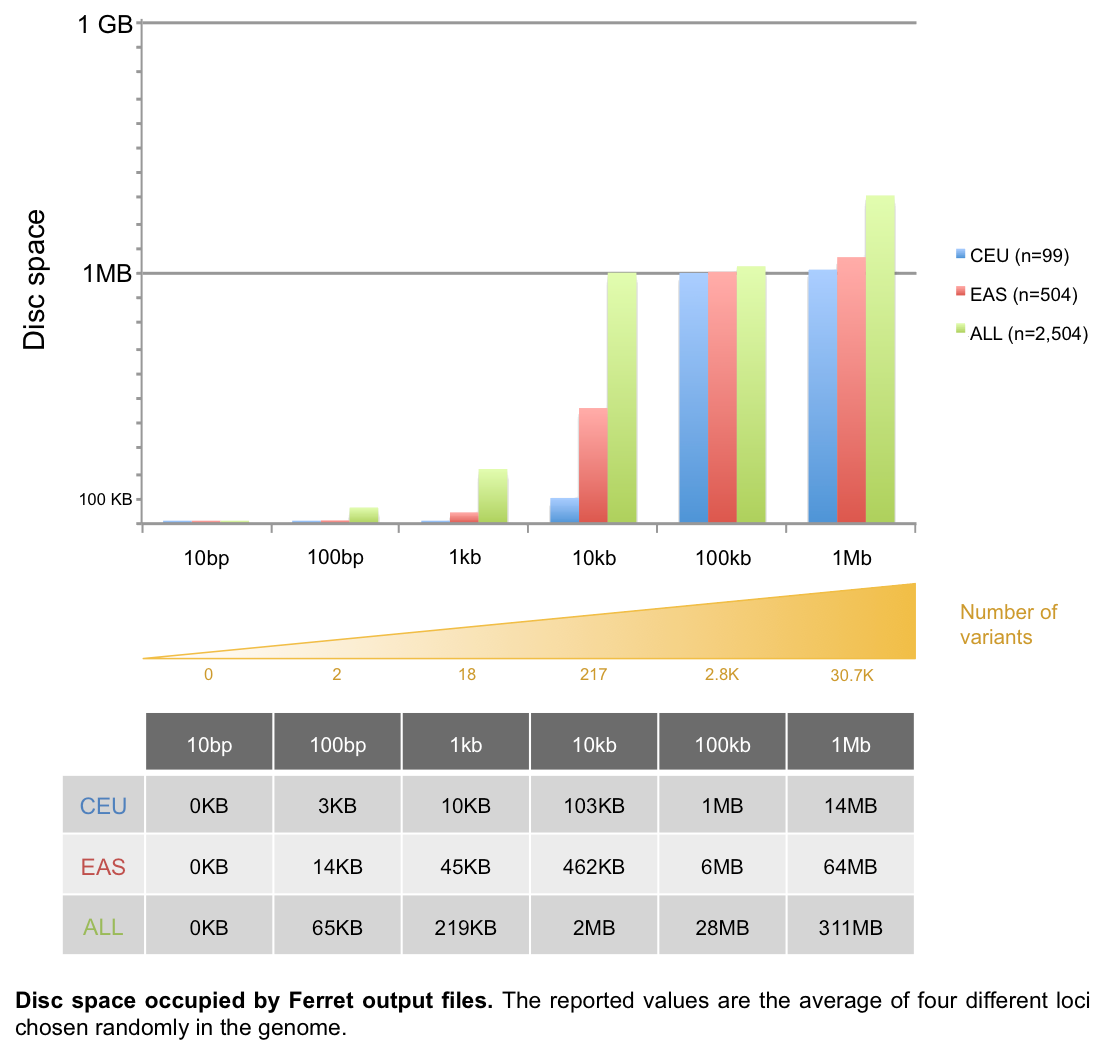
Ferret output files size
The reference population(s) size is the main factor impacting output file size:
- For a 100kb locus, 1MB disc space would be occupied for the CEU individuals while 28MB would be required for all 1KG population
- For a 1Mb locus, 14MB and 311MB disc space would be necessary for CEU and all individuals, respectively.
Contact
Developed by Sophie Limou and Andrew Taverner: ferret@nih.gov.
We encourage user community feedback to elicit new features.
Citation
Limou S, Taverner AM, Winkler CA. Ferret: a user-friendly Java tool to extract data from the 1000 Genomes project. Bioinformatics 2016, doi: 10.1093/bioinformatics/btw147 (View publication).
Download Ferret
Ferret’s only pre-requisite is to have Java JRE 8 installed on your computer. In addition, Ferret requires Java JDK 8 to retrieve allelic frequencies from the Exome Sequencing Project.
For queries from a Terminal console and for integration into pipelines, we also provide Ferret perl script and Java source code.
Version history
2016, November 23
Ferret was updated due to the recent NCBI security switch to HTTPS only.
2016, February 19
The usability and flexibility of Ferret were improved for this version:
- Clarification of input formats through tooltips,
- Implementation of a progress bar,
- Settings panel to select 1KG version, human genome version, and output format,
- Possibility to set a MAF threshold for variants to output.
2015, August 12
This version was presented at the ASHG 2015 meeting and includes several new features:
- Retrieves 1KG data from a locus, gene(s), or SNP(s) of interest,
- Displays allelic frequency for CNVs (in addition to SNPs and indels),
- Retrieves allelic frequency from the Exome Sequencing Project.
2013, October 23
This initial version corresponds to the version presented at the ASHG 2013 meeting, allowing to retrieve 1KG data from a locus of interest.
Future directions
We are currently working on the development of the following features:
- Parallelizing Ferret jobs to speed up runtime and increase the input size,
- Display variant annotations (e.g. functional consequences),
- Option to specify the 1000 Genomes .vcf URL.
FAQ
- How to report a bug or ask a question?
- Which Java version does Ferret require to run on my computer?
- Which Ferret version do I have on my computer? How do I get the latest Ferret update?
- Ferret is not working: nothing is happening when hitting the ‘Run’ button.
- How can I get Ferret to run on a Linux/Ubuntu environment?
- I’m getting an error when trying to retrieve allelic frequencies from the Exome Sequencing Project. Why?
- Why is my gene or my variant not retrieved by Ferret?
- How do I get the allelic frequencies in two different populations?
- How do I input more than one gene?
- How do I input more than one SNP?
- What 1000 Genomes version does Ferret query?
- What human genome version does Ferret expect?
- A lot of reported variants are monomorphic in my population(s) of interest. Can I set a minor allelic frequency (MAF) threshold?
- What can I do with the output files?
- In the .frq file, what do the 1KG_A1_FREQ and 1KG_A2_FREQ fields correspond to?
- In the .frq file, what do the ESP6500_EA_A1_FREQ and ESP6500_EA_A1_FREQ fields correspond to?
- Why do I get more SNPs in the output files than input in the SNP tab?
- Why do I get CNV information in the .frq file and not in the Plink/HaploView files?
- Why do the indels appear as A/T SNP in Plink/HaploView?
- Can Ferret generate data in a different output format?
- Why are the gene coordinates different from the 1000 Genomes browser?
1. How to report a bug or ask a question?
First, make sure that you have the latest version installed on your computer (see #2 and 3). Then, please check carefully the website and FAQ to be sure that your problem was not already solved. Finally, you can send us an email (ferret@nih.gov) and we’ll do our best to answer you in a timely manner. Please provide us with the following information so we can better assist you:
- - Your operating system and Java versions
- - Ferret eventual pop-up messages
- - Whether there is a temporary .vcf file created when you launch Ferret
- - Launch Ferret from a Terminal console (
java –jar Path/to/Ferret.jar), try to run your problematic query and provide us with any error message appearing in the Terminal console.
2. Which Java version does Ferret require to run on my computer?
Ferret’s only pre-requisite is to have Java JRE 8 installed on your computer. In addition, if you are interested in querying the Exome Sequencing Project, be sure to also install Java JDK 8. The Java installations require administrative privileges.
For Linux/Ubuntu users, the Java JRE/JDK installation can be tricky but you can find help online from the active Linux community (like here for Debian Linux, which includes Ubuntu).
3. Which Ferret version do I have on my computer? How do I get the latest Ferret update?
To find the version of Ferret installed on your computer, launch Ferret and the version number will appear in the window title (e.g. Ferret v2.1). You can also find the information in Help/About Ferret.
To check if a new Ferret version was released, go to Ferret/Check for updates: Ferret will check our website and invite you to download the latest version if necessary.
4. Ferret is not working: nothing is happening when hitting the ‘Run’ button.
Please make sure that your Internet connection is working properly, as Ferret needs to connect to the 1000 Genomes FTP and/or NCBI servers. Both servers are extensively accessed everyday all over the world, so please try again later to ensure that the problem does not come from overuse of NCBI and/or 1000 Genomes servers, rather than from Ferret itself. If the problem persists, do not hesitate to contact us (see #1).
5. How can I get Ferret to run on a Linux/Ubuntu environment?
If double-clicking the Ferret.jar file does not work, launch Ferret from a terminal window by typing the following command line: java –jar Path/to/Ferret.jar. If the problem persists, do not hesitate to contact us (see #1) and along with your Linux distribution, indicate the outputs generated from the following commands: java –version and javac –version.
6. I’m getting an error when trying to retrieve allelic frequencies from the Exome Sequencing Project. Why?
Be sure that you have Java JRE and JDK installed (see #2), and that you haven't renamed the Ferret application. If the problem persists, do not hesitate to contact us (see #1) and indicate whether a temporary file called evsClient0_15.jar is created in the same folder Ferret_vX.jar is installed in.
7. Why is my gene or my variant not retrieved by Ferret?
Ferret cannot currently report data from the sexual or mitochondrial chromosomes. Also, be sure to check that your gene or variant of interest is annotated on the human genome version being queried (hg19/GRCh37 by default).
8. How do I get the allelic frequencies in two different populations?
The allelic frequencies information is summarized in the .frq file (see #14).
If you are interested in the allelic frequencies in the combined group of two 1000 Genomes reference populations (e.g. CEU + TSI), just select simultaneously the two populations as input.
If you want to access the data from two populations independently (e.g. CEU and TSI separately), you will have to run two independent queries in Ferret (aka, first on CEU, and second on TSI).
9. How do I input more than one gene?
You have two options:
- 1. You can enter manually the list of genes separated by a comma in the input box;
- 2. You can load a comma-delimited .csv, a tab-delimited .tab or .tsv, or a space-delimited .txt file containing the list of genes. A carriage return can also be used as a delimiter for all file types.
Important: do not forget to indicate if your input corresponds to gene name or gene ID.
10. How do I input more than one SNP?
You have two options:
- 1. You can enter manually the list of SNPs separated by a comma in the input box;
- 2. You can load a comma-delimited .csv, a tab-delimited .tab or .tsv, or a space-delimited .txt file containing the list of SNPs. A carriage return can also be used as a delimiter for all file types.
Important: indicate only the unique rs number without the letters ‘rs’ (e.g. 2395029 for rs2395029).
11. What 1000 Genomes version does Ferret query?
By default, Ferret queries the most recent 1000 Genomes Phase 3. Alternatively, the user can query the Phase 1 release (go to Ferret/Settings).
12. What human genome version does Ferret expect?
By default, Ferret expects hg19/GRCh37 human genome version coordinates, as stated in the locus tooltip. Alternatively, the user can opt for the hg38/GRCh38 genome coordinates to query the 1KG Phase 3 release (go to Ferret/Settings).
Note: The Exome Sequencing Project data have not been converted to the hg38/GRCh38 genome version so far.
13. A lot of reported variants are monomorphic in my population(s) of interest. Can I set a minor allelic frequency (MAF) threshold?
Yes, you can set-up Ferret to only output variants with a frequency above a certain threshold (go to Ferret/Settings). The MAF threshold is applied to the selected 1KG populations. Alternatively, you can also apply the MAF threshold to the Exome Sequencing Project populations: variants respecting the MAF threshold in the selected 1KG populations, or in the Exome Sequencing Project African American or European American populations will then be printed out.
14. What can I do with the output files?
Ferret generates 4 output files:
- .frq: list of all identified variants (SNPs, indels and CNVs) along with chromosome, variant ID, hg19 chromosome position and alleles, as well as allelic frequency in the selected population(s) (see also #15 and #16). The variant ID corresponds to the rs number when available, and to the hg19 chromosome position when no rs number is reported (see also #19).
Note: this file can be open with a text editor program (e.g. Notepad) or with a spreadsheet program (e.g. Excel). - .map
- .ped
- .info
The .map, .ped and .info files allow you to open, visualize and manipulate the genotype data with the popular Plink and HaploView tools. You can then easily access linkage disequilibrium and haplotype information for the 1KG SNPs and indels, merge the 1KG data with your own datasets, design tagSNPs, etc.
15. In the .frq file, what do the 1KG_A1_FREQ and 1KG_A2_FREQ fields correspond to?
For each variant, the 1KG_A1_FREQ and 1KG_A2_FREQ fields correspond to the allelic frequency in the input population(s) for allele 1 and allele 2, respectively.
If you have selected to output the allelic frequencies from the Exome Sequencing Project (ESP) as well (see #16), only the 1KG_A1_FREQ field will appear in the .frq file.
16. In the .frq file, what do the ESP6500_EA_A1_FREQ and ESP6500_EA_A1_FREQ fields correspond to?
These fields only appear in the .frq file if you selected to output Exome Sequencing Project (ESP) frequencies. For each variant, the ESP6500_EA_A1_FREQ and ESP6500_AA_A1_FREQ fields correspond to allele 1 frequency in the European American and African American ESP6500 datasets, respectively. The last ESP release is based on 6500 US individuals, including 4300 European Americans and 2203 African Americans (for more details, see the ESP website). ESP only provides allele and genotype counts, and does not provide genotype data: as a consequence, the Plink/HaploView files only contain the 1000 Genomes Project data.
17. Why do I get more SNPs in the output files than input in the SNP tab?
If you input a variant, it is possible to get more than one variant in the output files if your SNP of interest is overlapping with another variant (e.g. indel or CNV). Alternatively, you can get more variants if you elect to include surrounding variants in a certain base-pair window range around your SNP of interest.
18. Why do I get CNV information in the .frq file and not in the Plink/HaploView files?
Plink and HaploView do not support CNV data. However, we included CNV information in the .frq file, as it can be useful when studying a region of interest (e.g. for primer design).
19. Why do the indels appear as A/T SNPs in Plink/HaploView?
Plink and HaploView do not support indels. We transformed the small indels into A/T SNPs so Plink and HaploView can read them. To get the allele correspondence, report to the variant ID: the first allele corresponds to A when the second allele corresponds to T (e.g. indel_rs136158_AT/A for rs136158, where the first allele AT has been transformed in A, and the second allele A has been transformed in T).
The indel representation follows the vcf format coding where AT/A corresponds to a T/- deletion, and C/CA corresponds to a -/A insertion.
20. Can Ferret generate data in a different output format?
If you are not interested in the genotype data, Ferret can generate the .frq file only.
For more advanced users, we also offer to output the 1KG data in the .vcf format, which can then be merged with the user’s dataset, or used to filter out specific variants using VCFtools, or to annotate variants (e.g. gene location, functional prediction, GWAS catalog) using Annovar. Note: the Exome Sequencing Project does not provide genotype data and hence cannot be included in the .vcf output.
At this stage, Ferret does not offer other output formats. However, if you are interested in the popular binary Plink (.bed/.bim/.fam) or Oxford (.gen/.sample) genomic file format, we recommend converting them from our default Plink outputs using the --make-bed Plink option and the -P GTOOL option.
21. Why are the gene coordinates different from the 1000 Genomes browser?
For gene coordinates, Ferret queries the curated and non-redundant NCBI’s RefSeq database, when the 1000 Genomes browser is based on the Ensembl human gene annotations. The coordinates reported in these two databases are greatly overlapping, however some minor discrepancies can still occur.